
MEET US AT THE IBERIAN PEPTIDE MEETING

Join us at the17th Iberian Peptide Meeting to be held in Madrid, Spain, on February 5-7, 2020.
Bachem supports its customers in the pursuit of groundbreaking discoveries that further scientific advances, particularly in the field of medicine. With a record of accomplishment in custom synthesis projects, the high quality of our peptides for research and development projects and our capacity to upscale the production of simple and modified peptides, we are the Pioneering Partner for Peptides.
The Bachem team at EPI is excited to meet with you, learn your needs for peptides and discuss how Bachem can assist to advance your research. We invite you to drop by our booth or to contact us to schedule a meeting in advance.
We look forward to meeting you at the 17th Iberian Peptide Meeting in Madrid!
CLICK CHEMISTRY
Bachem highlights the importance of «click reactions» in peptide chemistry as a simple and versatile concept for peptide synthesis and chemoselective modification. The broad spectrum of applications of the reaction includes ligation, cyclization, bioconjugation, and radiolabeling of peptides.
«Click Chemistry» is a term introduced by K.B.Sharpless, H.C.Kolb, and V.V.Fokin from the Scripps Research Institute at La Jolla to describe chemistry tailored to generate substances quickly and reliably by joining small units together similar to the modular strategy adopted by Nature. The term “click chemistry” applies to reactions that are highly efficient, wide in scope, and stereospecific. Product isolation is easy, the reactions are simple to perform using inexpensive reagents and can be conducted in benign solvents such as water.
The Huisgen 1,3-dipolar cycloaddition is probably the most extensively studied click reaction. A variant of this reaction, the copper-catalyzed azide-alkyne cycloaddition (CuAAC), independently developed by the groups of Sharpless at Scripps and Morten Meldal at Carlsberg Laboratory in Denmark, fits the «click chemistry» concept well and is one of the most popular prototype click reactions to date.
Chemistry of CuAAC
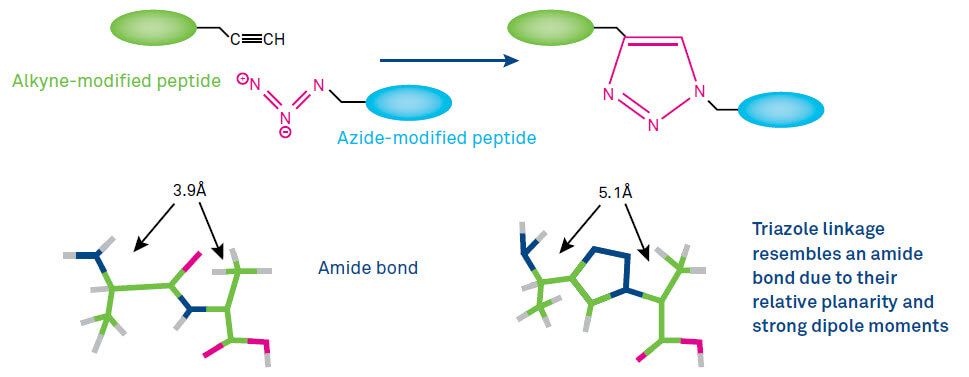
The popularity of the CuAAC is largely a result of the unique properties of both azides and the resulting triazoles. CuAAC involves the formation of a 1,2,3-triazole ring which is a rigid five-membered heterocycle. Such triazoles are isosteres of the peptide bond, mimicking the planarity of the amide moiety, but less prone to hydrolytic cleavage (Figure 1).
Most click reactions involve carbon-hetero atom bonding processes and have a high energy content which make the additions irreversible. Furthermore, azide moieties are easy to introduce, stable to water and oxidative conditions, orthogonal to many commonly used functional groups and vigorously reactive with others. For applications in vitro and in vivo, azides are virtually absent from any naturally occurring species (bioorthogonal). The combination of the robustness of the triazole bond, the resemblance to an amide bond, and the potential biological properties it could endow make the triazole linkage not merely a benign, easily synthesized linker, but an integral part of the success of click chemistry.
Figure1: Clicking of an alkyne-functionalized peptide with an azide-modified substrate in the presence of Cu(I) to form a triazole-linked conjugate.
Click Chemistry Involving PeptidesClick chemistry provides a number of avenues for peptide or protein modifications and could be combined with other techniques to make complex structures and multi-component functionalized systems with ease. The chemistry can be performed in different ways. For example, peptides can be converted post-synthetically to an azido derivative which can be clicked with appropriate substrate containing a clickable alkynyl group or vice versa. Peptides can also be made by inter- and intramolecular click reactions using azide or alkyne containing amino acids or building blocks during peptide synthesis. Building blocks containing clickable moieties will be instrumental for constructing side-chain modified peptides, interside-chain peptide chimera, peptide small molecule conjugates, and cyclic peptides. Solid phase resins modified with clickable groups can also be used for making clickable or modified peptides. Click chemistry is compatible with various protected amino acid side chains used in peptide synthesis.
A number of reagents and building blocks can be used for click chemistry. These include ω-azido-α-amino acids, PEG and spacer azides and alkynes, azide- or alkyne-modified fluorescent dyes and quenchers, nucleosides and nucleotides, alkyne or azide-containing chemical modification reagents, diazo transfer reagents, and propargyl derivatives of amino acids (e.g.O-propargylserine, glutamic acid bispropargyl amide)
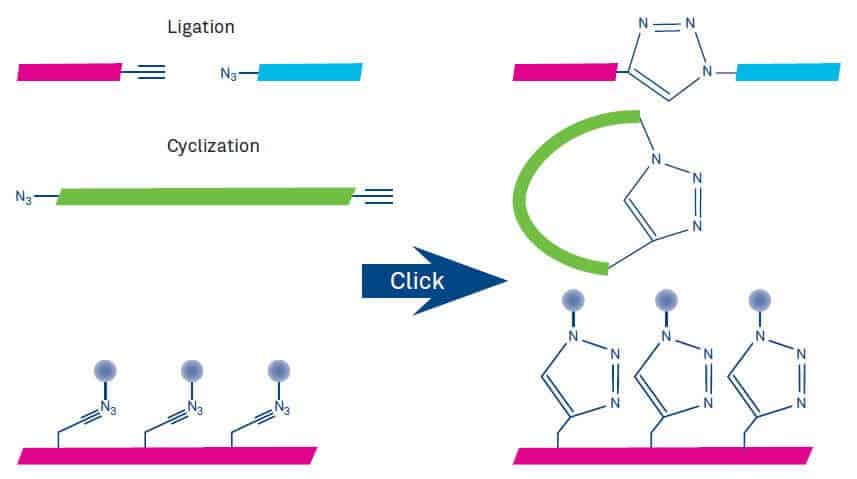
The most important applications of click chemistry in peptide science include chemical ligation, cyclization and bioconjugation (Figure 2). Other typical applications are conjugation of isotope labels for imaging, synthesis of peptidomimetics based on the triazole backbone, conformational and backbone modifications.
Figure 2: Utilization of click chemistry for ligation, cyclization and conjugation of peptides.
Chemical Ligation and Peptide Modification
Linking two or more peptide fragments together to make a larger peptide chain is called ligation. Click chemistry can be conveniently utilized to make peptide–peptide linkages. A peptide fragment functionalized with an alkyne group could be ligated to another peptide with an N-terminal azide moiety resulting in a triazole linker (similar to an amide bond as explained earlier) holding two peptide units together. Similarly multimeric peptides can be obtained by incorporating orthogonal side chain protecting groups such as ivDde or Aloc (for modifying the side chain of Lys) followed by selective deprotection, attachment of an alkyne function and clicking with N-terminal azide peptides. Numerous examples of peptide ligation have been published such as
synthesis of a clickable RGD peptide (obtained by reacting Lys side chain with azido acetic acid) that can be linked to another peptide fragment
- synthesis of a cell-permeable peptide therapeutic by clicking the alkynyl-modified peptide drug (using inexpensive propargylamine or 1-(2-nitrophenyl)propargyl alcohol) with nona-arginine modified with an azide group
- synthesis of neurotensin (8-13)-containing heterodimers by clicking alkyne-neurotensin (obtained by reacting with succinimidyl-hex-5-ynoate, the NHS ester of Bachem product 4066756) with the azide of a Plk1-PBR binding phosphorylated hexapeptide (made by reacting with succinimidyl-4-azidovalerate, the NHS ester of Bachem product 4059927). The resulting triazole-containing oligopeptides were found to self-dimerize in a head-to-tail fashion as the native peptides.
Modification of peptides by PEGylation has been achieved by click chemistry. For example, a lipopeptide was assembled by solid-phase synthesis followed by an on-resin PEGylation reaction (using azido-PEG) and cleavage of the PEGylated peptide from the resin. There is a tremendous potential for click chemistry for various chemical modifications of peptides and proteins (e.g. attaching ligands, liphophilic or hydrophilic groups or linkers etc.).
1. Peptide Cyclization:
A variety of macrocyclization methods are available to increase the clinical efficacy and bioavailability of peptides. The click reaction has been exploited in a number of different cyclization reactions such as the on-resin cyclization of a disulfide-containing peptide before or after removal of the side-chain protecting groups; the preparation of novel heterodetic cyclopeptides containing a triazole bridge by an intramolecular side chain-to-side chain click reaction; Cu(I)- and Ru(II)-mediated click cyclizations of tripeptides for generating vancomycin-inspired mimics; on-resin cyclization of peptide ligands of the vascular endothelial growth factor (VEGF) receptor-1 etc.
In many cases, formation of considerable amounts of macrocyclic heterodimers was observed during click-mediated macrocyclization reactions, opening up the prospects of synthesizing complex peptide structures, which are otherwise difficult to obtain. A novel stapling methodology for 310-helical peptides using CuAAC click reaction in a model aminoisobutyric acid (Aib)-rich peptide resulted in a more ideal 310 helix than its acyclic precursor.
2. Bioconjugation:
Bioconjugation is the process by which synthetic molecules are attached to biological targets, or by which biomolecules are linked together. The impact of click chemistry on bioconjugation has been extensive in recent years. Arginine-rich TAT peptides modified with a clickable azido group can be conjugated to oligonucleotides, cytotoxic drugs, kinase inhibitors etc. to facilitate cell penetration for therapeutic purposes. The application of the CuAAC reaction provides a powerful chemical method to access mimetics of glycopeptides and glycoproteins (neoglycopeptides and neoglycoproteins) of well-defined homogeneous structure. Complex cyclopeptide-centered multivalent glycoclusters has been synthesized using the Cu-catalyzed click reaction. Self-assembling peptide fibers can incorporate multiple clickable peptides non-covalently, stoichiometrically and without disrupting their structure or stability. They can be conjugated to biotin followed by binding of streptavidin-nanogold particles, or rhodamine, and visualized by electron and light microscopy. This approach allows the development of multi-component functionalized systems. The click reaction allows conjugating fluorescent molecules to peptides and proteins under mild conditions, a most important application in the emerging field of cell biology and functional proteomics.
3. Peptidomimetics Design, Synthesis and Drug Discovery:
The triazole linkage has found particularly broad use in the field of peptidomimetics. The triazole unit is resistant to enzymatic degradation, hydrolysis, and oxidation, making it an attractive moiety to replace more labile linkers in biologically active compounds. The click reaction has been utilized as a conjugation strategy in the design and synthesis of complex biomimetic architectures in which the triazole linkage replaces, and in some cases acts as a surrogate for peptide and phosphodiester bonds. Replacing a peptide bond with a triazole unit could result in interesting structures with unique conformational characteristics. Triazole units formed by the click reaction can act as helical component, a β-turn unit and a cis/trans-prolyl ratio modifier. Triazole units can also act as an effective replacement for a peptide portion in HIV-1 protease inhibitors. Modified peptides in which a triazole ring is introduced in the peptide backbone or attached to the side chain of a residue are good candidates to design new antimicrobial agents.
4. Radiolabeling and Imaging:
The CuAAC is an ideal ligation reaction for radiolabeling sensitive biomolecules.
Alkyne or azide derivatives of radioisotope-containing compounds could be used for labeling biomolecules such as folic acid, peptides, proteins, and glycopeptides. For example, an 11C isotope label was introduced via converting [11C]-CH3I into [11C]-CH3N3 by nucleophilic substitution and subsequently reacting the azide with an alkyne-modified peptide. 18F labeling for PET imaging was achieved by clicking azidomethyl-4-[18F]-fluorobenzene to a modified peptide.
An important limitation of CuAAC exists for chelators like DOTA, since they form a complex with the catalyst. Therefore, conjugates with such compounds are more difficult to obtain.
Cu-free Click Reactions
The cytotoxicity of copper remains a concern and a limiting factor for the widespread in vivo application of the CuAAC reaction.
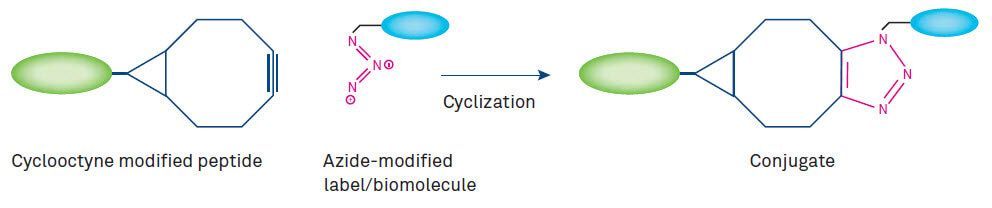
Meanwhile, Cu-free alternatives have been developed. Copper-free click chemistry is based on the reaction of strained cyclooctynes (such as BCN, DBCO) or cyclooctynes activated by electron-withdrawing substituents (MOFO, DIFO) with azides in the absence of Cu catalyst at low temperature. The SPAAC (strain-promoted alkyne-azide click chemistry) reaction developed by Carolyn Bertozzi’s group can be applied for in vivo chemoselective ligation to biomolecules in the same manner as the Staudinger ligation (reaction between a phosphine and an azide with release of nitrogen), but with the advantage of a much more rapid reaction. Recent applications of Cu-free click chemistry to peptides include the synthesis of a DOTA-peptide conjugate prepared by the attachment of DOTA to MOFO followed by conjugation to an azide-modified α-MSH peptide. The resulting conjugate can form chelates with radionuclides for imaging applications such as tumor targeting.
As the cyclooctyne moieties vary considerably in reactivity, multiple SPAAC is feasible.
Figure 3: Copper-free click reaction using cyclooctyne-based substrates.
References
CLICK CHEMISTRY APPLIED TO DRUG CONJUGATES IN CLINICAL DEVELOPMENT
Click chemistry has been increasingly utilized in the synthesis of Antibody-Drug-Conjugates (ADCs). ADCs combine the specificity of antibodies for tumor targeting with highly cytotoxic small molecules through chemical linkers. First generation ADCs such as Genentech’s Polivy are mixtures of many different biologic conjugates due to the inability of traditional coupling reactions to control how many drug molecules conjugate to the monoclonal antibody (mAb) or to which position within the mAb. With click chemistry, a bioorthogonal handle on the mAb provides a way to control the drug-antibody ratio (1). This method is being used by companies to create a new generation of ADCs and some of these ADC candidates are already in Phase I clinical trials as shown in Table 1.
| Product Name | ADC Components | Pipeline Indication(s) | Highest Phase | Companies (Partner) |
|---|---|---|---|---|
| ARX788 | HER2 targeting antibody + Amberstatin269 | Ovarian Cancer, Esophageal Cancer, Adenocarcinoma Of The Gastroesophageal Junction, Pancreatic Cancer, Gastric Cancer, Metastatic Breast Cancer, Colon Cancer, Gastroesophageal (GE) Junction Carcinomas | Phase I | Ambrx; Zhejiang Medicine |
| TRPH-222 | Anti-CD22 antibody + maytansine | Follicular Lymphoma, Acute Lymphocytic Leukemia (ALL, Acute Lymphoblastic Leukemia), Diffuse Large B-Cell Lymphoma, Mantle Cell Lymphoma, Marginal Zone B-Cell Lymphoma | Phase I | Catalent/ Redwood Bioscience (Triphase Accelerator Corp) |
| STRO-001 | Anti-CD74 IgG1 antibody (SP7219) + maytansine | Follicular Lymphoma, Diffuse Large B-Cell Lymphoma, Mantle Cell Lymphoma, Refractory Multiple Myeloma, Relapsed Multiple Myeloma | Phase I | Sutro Biopharma |
| STRO-002 | Anti-FolRa human IgG1 antibody (SP8166) + 3-aminophenyl hemiasterlin | Fallopian Tube Cancer, Epithelial Ovarian Cancer, Endometrial Cancer, Peritoneal Tumor | Phase I | Sutro Biopharma |
| ADCT-601 | Anti-AXL IgG1 antibody + Pyrrolobenzo-diazepine dimer SG3199 | Breast Cancer, Ovarian Cancer, Renal Cell Carcinoma, Non-Small Cell Lung Cancer, Esophageal Cancer, Malignant Mesothelioma, Soft Tissue Sarcoma, Colorectal Cancer, Head And Neck Cancer, Pancreatic Cancer, Gastric Cancer | Phase I | Synaffix (ADC Therapeutics |
Clinical Drug Candidates
Ambrx is developing ARX788, a HER2 specific monoclonal antibody conjugated with Amberstatin269, a potent cytotoxic tubulin inhibitor. ARX788 utilizes site-specific conjugation technology. Zhejiang Medicine has licensed the commercial rights to ARX788 in China. In 2019, the two companies presented positive top line data from a Phase Ia/Ib clinical trial of ARX788 in metastatic HER2 positive breast cancer. An overall response rate of 63% was observed at 1.5mg/kg. ARX788 was well tolerated with no established Dose Limiting Toxicity or Maximum Tolerated. (2)
TRPH-222 is a site-specific ADC under development by Triphase Accelerator Corp. This product candidate is a modified humanized antibody conjugated to a maytansine payload using Hydrazino-Pictet-Spengler (HIPS) chemistry and a proprietary 4AP linker. TRPH-222 targets cells expressing CD22 and uses the SMARTag® platform. In 2019, Triphase Accelerator initiated a Phase I clinical trial of TRPH-222 in patients with relapsed and/or refractory B-cell lymphoma (2).
Sutro Biopharma is developing STRO-001 for the treatment of B-cell malignancies such as multiple myeloma and diffuse large B-cell lymphoma. The drug candidate is a monoclonal antibody conjugate that targets cells expressing CD74. Sutro’s cell-free protein synthesis and site-specific conjugation platform technology was used to discover and develop STRO-001. In 2019, Sutro announced positive interim Phase I safety data from a Phase I dose escalation trial of STRO-001 (2).
Sutro Biopharma is also developing STRO-002, a monoclonal antibody conjugate that acts by targeting folate receptor alpha (FolRα) (3). STRO-002 is a drug candidate for the treatment of relapsed/refractory cancers like epithelial ovarian cancer, endometrial carcinoma, fallopian tube cancer and peritoneal cancer. In 2019, Sutro Biopharma initiated a Phase I clinical trial of STRO-002 in patients with ovarian and endometrial cancers (2).
ADCT-601 is under development by ADC Therapeutics for the treatment of cancers such as relapsed and refractory colorectal cancer, esophageal cancer, and gastric cancer. ADCT-601 is an ADC conjugated using using Glycoconnect™ technology (licensed from Synaffix BV) and it targets cells expressing tyrosine protein kinase receptor UFO, an enzyme encoded by the AXL gene. In 2019, ADC Therapeutics announced the initiation of a Phase I clinical trial to evaluate the safety, tolerability, pharmacokinetics and anti-tumor activity of ADCT-601 in patients with advanced solid tumors (2).
Conclusion
ADCs have emerged as a promising drug class in the field of oncology medicine. Click chemistry is playing an important role in constructing ADCs with defined and uniform sites of drug conjugation.
References
1) M. Peplow, Click chemistry targets antibody-drug conjugates for the clinic. Nature Biotechnology 2019, 37, 835-837.
2) GlobalData. 2019
3) Sutro Biopharma initiates Phase I clinical trial of STRO-002 for the treatment of ovarian and endometrial cancers. Sutro Biopharma 2019
MEET BACHEM: MANUELA MEISTER
What is your official job title at Bachem?
My job title is Senior Chemist QC.
How long have you been with Bachem? Where did you work before Bachem?
I have been working at Bachem for 2 years now. I started as Project Chemist and became later Senior Chemist. Before Bachem, I did my Master’s Degree in Physical Chemistry at the ZHAW.
Briefly, what do you do at Bachem?
I work in the QC Late Phase department and I am responsible for method development (GC, IC, IR), qualification of instruments, supporting at troubleshooting, organizing and prioritizing different analysis and for the release of raw and starting materials.
What is your academic background/degrees or training?
The topic of my bachelor’s degree in nanotechnology was the synthesis and analysis of phosphorescent nanoparticles. The topic of my master’s degree in physical chemistry was the enantiomeric separation of small chiral molecules.
What do you like to do outside of work?
I enjoy spending my time in nature, being with my family and friends, doing sport (fitness, bouldering, skiing, snowboarding), cooking and travelling.
What do you like most about your job?
I like being involved in different projects, which makes my job very interesting and versatile. Besides that, I appreciate getting in contact with many different people from in- and outside of Bachem.
Thank you very much Manuela.

Peptide highlights
Interesting news about peptides in basic research and pharmaceutical development:
Newly engineered peptide shows potential as long-acting anti-HIV drug-Science Daily
Is a universal vaccine against cancer possible?-Drug Target Review
SMART and NTU researchers design polymer that can kill drug-resistant bacteria-MIT News
Peptide catalyzes macrocycle formation-Chemical & Engineering News
LITERATURE CITATIONS
Bachem peptides and biochemicals are widely cited in research publications. Congratulations to all our customers with recent publications!
K.Gupta et al.
Exploring structural dynamics of a membrane protein by combining bioorthogonal chemistry and cysteine mutagenesis.
C.Rimbault et al.
Engineering selective competitors for the discrimination of highly conserved protein-protein interaction modules.
Nature Communications 10, 4521 (2019)
M.A.Betancourt-Solis et al.
The atlastin membrane anchor forms an intramembrane hairpin that does not span the phospholipid bilayer.
Journal of Biological Chemistry 293, 18514-18524 (2018)
W.Wu et al.
CTGF/VEGFA-activated fibroblasts promote tumor migration through micro-environmental modulation.