Amyloid Peptides Offered by Bachem
Extracellular amyloid-β peptide deposition into cerebellar plaques and formation of intracellular neurofibrillary fibers accompanied by the loss of neurons are characteristic histopathological lesions found in the brains of Alzheimer‘s disease patients. Individuals suffering from this disease show a gradual loss of cognitive functions and disturbances in behavior. Apart from some rare familial forms of the disease, the onset of Alzheimer‘s disease is usually above 60 years. Since the risk to develop the disease increases with age, Alzheimer‘s disease has turned into a major health and social problem in “first world” countries with an increasing proportion of older people, and is going to become one in emerging states. In this brochure we present amyloid peptides and related products for Alzheimer‘s disease research.
Alzheimer’s Disease
Alzheimer‘s disease (AD) is the prevalent cause of dementia in elderly people and has become one of the leading causes of death in developed countries together with cardiovascular disorders, cancer, and stroke. It is estimated that more than 46 millions of people suffer from AD all over the world. As age advances, the risk for developing AD increases. The frequency of AD at the age of 60-64 is about 1% and doubles approximately every five years. By the age of 90 and older, approximately 50% of the population suffers from this disease. AD is an irreversible and progressive neurodegenerative disorder. Symptoms include gradual loss of cognitive functions such as memory, verbal and visuospatial abilities, changes in personality, behavior, and activities of daily living. AD patients in the final stages are completely dependent on the care of others.
Amyloid beta-protein
(1-42)
Cleavage of amyloid precursor protein (APP) by β- and γ-secretases yields amyloid β peptides. Aβ 1-40 and the more virulent Aβ 1-42 are the most important APP degradation products. Aβ42 is the main constituent of amyloid plaques.
The characteristic lesions in the brains of AD patients were first described by the German neuropsychiatrist Alois Alzheimer in 1906 during the post-mortem examination of a mentally ill patient whose deterioration he had observed until her death. The lesions consisted of dense extracellular deposits, now designated as neuritic or senile plaques, and intracellular dense bundles of fibrils, which are now known as neurofibrillary tangles.
Currently, diagnosis of AD with adequate testing is approximately 90% accurate. It is based on the exclusion of a variety of diseases causing similar symptoms and a careful neurological and psychiatric examination, as well as neuropsychological testing. Imaging technologies for detecting amyloid plaques and tangles in vivo are becoming more precise and thus a valuable additional tool. Numerous potential biomarkers as α1 -antitrypsin, complement factor H, α2 -macroglobulin, apolipoprotein J, and apolipoprotein A-I for diagnosing AD are being evaluated. However, post-mortem histopathological examination of the brain is still the only definite diagnosis of this disease.
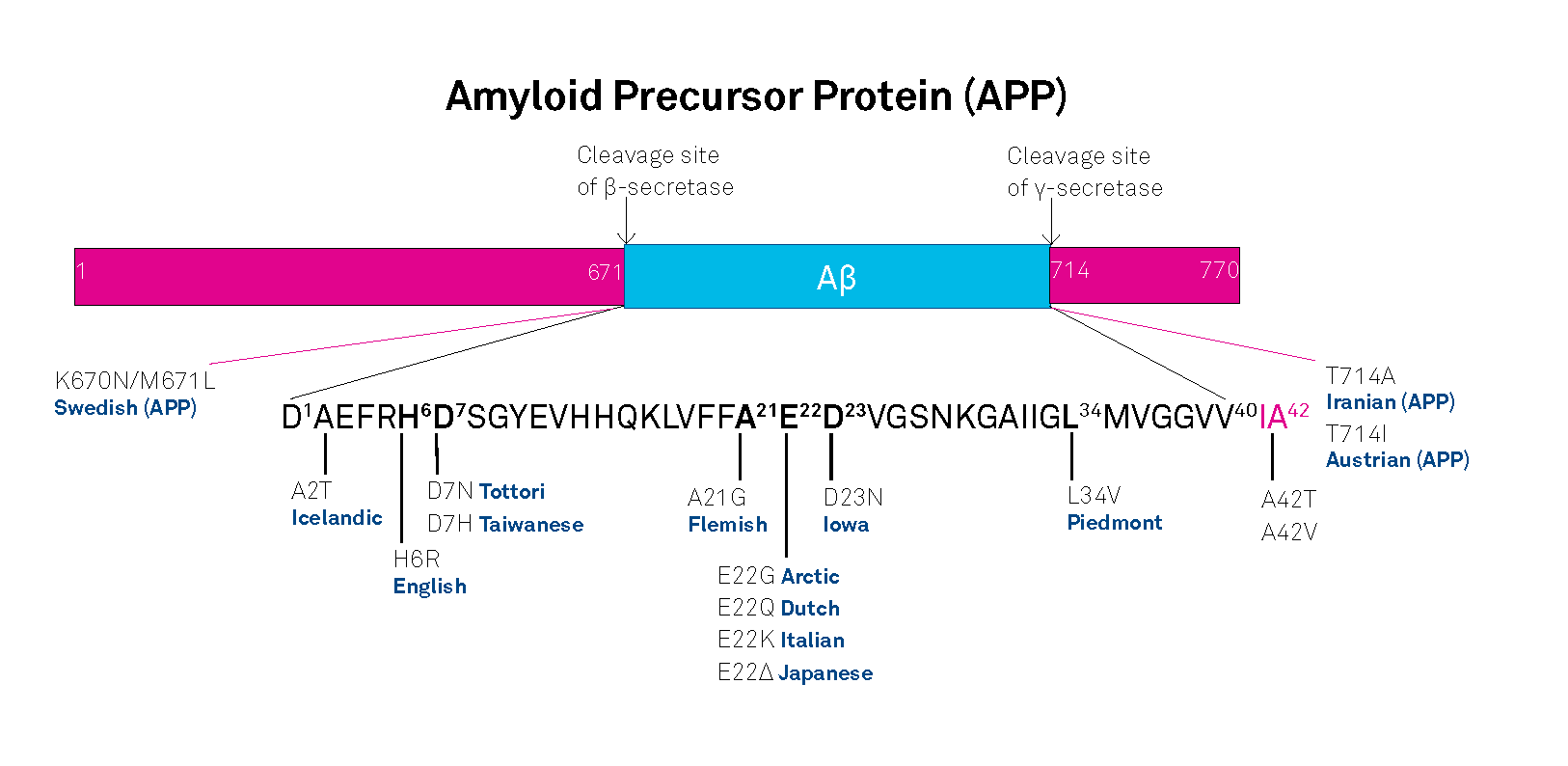
AD can be either inherited or sporadic. The inherited or familial AD is rare and comprises only 5-10% of all cases. Autosomal dominant mutations in the amyloid β/A4 protein precursor (APP) gene on chromosome 21 and the presenilin-1 or -2 genes on chromosomes 14 and 1, respectively, have been attributed to the early onset (before the age of 65) of this disease.
APP belongs to the type-1 integral membrane glycoproteins with at least 10 isoforms generated by alternative splicing of the 19 exons. The predominant transcripts are APP695, APP751, and APP770. A number of mutations within the APP gene have been detected in families with an inherited risk for early onset of AD. Usually, they are named after the region, in which they have been detected, e.g. the London APP717 mutations (V717I, V717F, V717G), the Swedish APP670/671 double mutation (K670N/M671L), the Flemish APP692 mutation (A692G), or the Dutch APP693 mutation (E693Q). The Swedish mutation of the β-secretase cleavage site of APP and mutations of positions 692-694 (Aβ 21-23), which strongly influence the aggregation behavior of Aβ, have been studied intensively.
A choice of relevant mutations in the Aβ region of APP is assembled in the table below.
| Exchanged Position in APP | Exchanged Position in Aβ | Designation |
|---|---|---|
| A673T | A2T | Icelandic |
| H677R | H6R | English |
| D678H | D7H | Taiwanese |
| D678N | D7N | Tottori |
| A692G | A21G | Flemish |
| E693D | E22∆ | Osaka |
| E693G | E22G | Arctic |
| E693Q | E22Q | Dutch |
| E693K | E22K | Italian |
| D694N | D23N | Iowa |
| L705V | L34V | Piedmont |
The presenilins are another group of proteins involved in the development of AD. Presenilins are integral membrane proteins with eight transmembrane domains localized in the endoplasmic reticulum and the Golgi apparatus. A multitude of mutations within the presenilin-1 and two within the presenilin-2 gene account for most of the cases of early onset of AD.
Genetic factors may contribute as well to the late onset of AD. Increased susceptibility is associated with the expression of different apolipoprotein E (ApoE) isoforms due to the polymorphism in the APOE gene on chromosome 19. In the central nervous system, ApoE has been implicated in growth and repair during development or after injury. Carriers of the APOEε4 allele show a higher risk in developing the disease than carriers of the other two possible alleles APOEε2 and APOEε3. The ApoEε4 effect seems to be dose-dependent since individuals with two of these alleles seem to be at two-fold higher risk to develop the disease than those with one allele. Polymorphisms of the α2 -macroglobulin gene on chromosome 12 and the gene coding low-density lipoprotein receptor-related protein 1 (LRP1), LRP1-C/T, have also been suggested to be a risk factor for the late onset of AD. However, further studies in this field are required.
A number of additional, most diverse risk factors have been proposed. These include gender, ethnic group, head trauma, cardiovascular diseases, and educational level.
AD THERAPEUTIC STRATEGIES RELY ON DETAILED KNOWLEDGE OF THE MOLECULES INVOLVED
Women, Hispanics, individuals who have experienced a head trauma earlier in life, and persons who suffer from cardiovascular diseases appear to have a higher risk of developing the disease.
The etiology of AD is still not completely understood. Initial research focused upon determining the molecular structure of the senile plaques and the neurofibrillary tangles originally described by Alois Alzheimer. The main constituents of the senile plaques were identified as cleavage products of APP, designated as amyloid β-peptides (Aβ peptides).
Depending on the composition and the fraction of fibrillar to non-fibrillar forms of these amyloid peptides, several kinds of senile plaques can be distinguished. Three types of proteases, α-secretase, β-secretase (or β-site APP-cleaving enzyme, BACE), and γ-secretase are involved in APP processing. APP can either be processed by the α- and γ- or by the β- and γ-secretases. The major two amyloid peptides identified in senile plaques, amyloid β-protein (1-40) (Aβ40) and amyloid β-protein (1-42) (Aβ42), are generated by successive proteolysis of APP by β- and γ-secretases. Cleavage of APP by β-secretase results in the release of the extracellular N-terminal protein fragment known as soluble APP-β molecule (sAPP-β). Then, the membrane-retained APP is further processed within the transmembrane domain by γ-secretase to yield either Aβ40 or Aβ42. The formation of Aβ40 and Aβ42 is a normal process, and both peptides can be detected in the plasma and cerebrospinal fluid (CSF) of healthy subjects.
In most studies, similar concentrations of Aβ40 have been measured in the CSF of both healthy controls and AD patients. On the other hand, Aβ42 concentrations in the CSF of AD patients are significantly lower than in normal controls, probably reflecting an increased deposition as insoluble plaques.
The neurofibrillary tangles found inside neurons of Alzheimer’s brains are composed of paired helical filaments whose main components are hyperphosphorylated forms of tau, a microtubule associated protein involved in promoting microtubule assembly and stabilization. Self-assembly into paired helical filaments is believed to be a result of hyperphosphorylation due to either the increased activity of protein kinases or the decreased activity of phosphatases.
Several lines of evidence support the view that the accumulation of Aβ42 in the brain is a primary event in the development of AD. Increased cerebral Aβ production appears to be characteristic for all the mutations within the APP and the presenilin genes of familial AD. In patients with Down syndrome (trisomy 21), elevated levels of APP and Aβ due to a third copy of the APP gene result in deposition of Aβ at an early age between 20 and 30.
Formation of neurofibrillary tangles is considered as a consequence of Aβ deposition with a further impact on the progression of the disease possibly due to disruption of axonal transport mechanisms in neurons.
The detailed knowledge about the molecules involved in AD has led to the development of several therapeutic strategies.
One strategy aims at the reduction of Aβ40 and Aβ42 by inhibition of either β- or γ-secretase activity or by clearance of Aβ in the brain by means of immunization with these peptides. Transition metals as Cu, Fe and Zn play an important role in the pathology of AD. Aggregation and neurotoxicity of Aβ are dependent on the presence of copper, so Cu-chelating agents showed promising effects in animal models. Another approach is the prevention of the cellular inflammatory response in the cerebral cortex elicited by the progressive accumulation of Aβ. Further preventive therapeutic strategies are based on the findings that cholesterol-lowering drugs such as statins and estrogen replacement therapy reduce the risk of developing AD. An additional treatment alternative would be the inhibition of the serine-threonine protein kinases, glycogen synthase kinase 3 (GSK3) and cyclin-dependent kinase 5 (CDK5), which are probably responsible for the phosphorylation of the tau protein. Inhibition of calpain, an enzyme showing increased activity in AD brains, led to promising results in animal studies. Calpain cleaves the CDK5 activator p35 leading to p25 formation and CDK5 overactivation.
Several acetylcholinesterase inhibitors such as tacrine, donepezil, rivastigmine, and galantamine have been approved for the treatment of mild to moderate AD by the FDA and other authorities. They act by reducing the deficits of the neurotransmitter acetylcholine associated with cognitive impairment in AD patients. The amantadine derivative memantine, an NMDA receptor antagonist, which was already used for the treatment of moderate to severe AD in Europe, has gained approval in the United States by the FDA as well.
A promising drug candidate, the β-secretase inhibitor verubecestat (MK-8931) developed for the management of mild to moderate AD, has moved to phase III. Moreover, the BACE inhibitor AZD3293 showed encouraging results in clinical studies. Antibodies as aducanumab and solanezumab, which have been designed to degrade plaques and lower the level of Aβ in the brain, have reached advanced stages of clinical testing for mild cases of AD.
Despite the many promising therapeutic approaches, AD still remains a major burden for the patients, their relatives, and the society.
Subscribe to our newsletter
"*" indicates required fields
References
P.M. Gorman and A. Chakrabartty
Alzheimer beta-amyloid peptides: structures of amyloid fibrils and alternate aggregation products.
Biopolymers 60, 381-394 (2001) Review
T. Hartmann
Cholesterol, A beta and Alzheimer‘s disease.
Trends Neurosci. 24, S45-S48 (2001) Review
P.L. McGeer and E.G. McGeer
Inflammation, autotoxicity and Alzheimer disease.
Neurobiol. Aging 22, 799-809 (2001) Review
D.J. Selkoe
Alzheimer‘s disease: genes, proteins, and therapy.
Physiol. Rev. 81, 741-766 (2001) Review
R. Cacabelos
Pharmacogenomics in Alzheimer‘s disease.
Mini Rev. Med. Chem. 2, 59-84 (2002) Review
J. Hardy and D.J. Selkoe
The amyloid hypothesis of Alzheimer‘s disease: progress and problems on the road to therapeutics.
Science 297, 353-356 (2002) Review
A.M. Palmer
Pharmacotherapy for Alzheimer‘s disease: progress and prospects.
Trends Pharmacol. Sci. 23, 426-433 (2002) Review
B.A. Vicioso
Dementia: when is it not Alzheimer disease?
Am. J. Med. Sci. 324, 84-95 (2002) Review
D.A. Butterfield and C.B. Pocernich
The glutamatergic system and Alzheimer’s disease: therapeutic implications.
CNS Drugs. 17, 641-652 (2003) Review
I. Churcher and D. Beher
gamma-Secretase as a therapeutic target for the treatment of Alzheimer‘s disease.
Curr. Pharm. Des. 11, 3363-3382 (2005) Review
E. Gazit
Mechanisms of amyloid fibril selfassembly and inhibition. Model short peptides as a key research tool.
FEBS J. 272, 5971-5978 (2005) Review
K. Irie et al.
Structure of beta-amyloid fibrils and its relevance to their neurotoxicity: implications for the pathogenesis of Alzheimer‘s disease.
J. Biosci. Bioeng. 99, 437-447 (2005) Review
M.R. Nichols et al.
Amyloid-beta aggregates formed at polar-nonpolar interfaces differ from amyloid-beta protofibrils produced in aqueous buffers.
Microsc. Res. Tech. 67, 164-174 (2005) Review
E.M. Sigurdsson
Amyloid Proteins – Methods and Protocols
Meth. Mol. Biol. 299, (2005)
A.K. Tickler et al.
The role of Abeta peptides in Alzheimer‘s disease.
Protein Pept. Lett. 12, 513-519 (2005) Review
P. Westermark
Aspects on human amyloid forms and their fibril polypeptides.
FEBS J. 272, 5942-5949 (2005) Review
E. Levy et al.
Studies on the first described Alzheimer’s disease amyloid beta mutant, the Dutch variant.
J. Alzheimers Dis. 9, 329-339 (2006) Review
K. Takano et al.
Structure of amyloid beta fragments in aqueous environments.
FEBS J. 273, 150-158 (2006)
T. Tomita and T. Iwatsubo
gamma-Secretase as a therapeutic target for treatment of Alzheimer‘s disease.
Curr. Pharm. Des. 12, 661-670 (2006)
Y. J. Wang et al.
Clearance of amyloid-beta in Alzheimer’s disease: progress, problems and perspectives.
Drug Discov. Today 11, 931-938 (2006)
J.X. Chen and S.D. Yan
Pathogenic role of mitochondrial amyloid-beta peptide.
Expert Rev. Neurother. 7, 1517-1525 (2007) Review
M.A. Findeis
The role of amyloid beta peptide 42 in Alzheimer’s disease.
Pharmacol. Ther. 116, 266-286 (2007) Review
V.H. Finder and R. Glockshuber
Amyloid-beta aggregation.
Neurodegener. Dis. 4, 13-27 (2007) Review
M. Li et al.
The role of intracellular amyloid beta in Alzheimer’s disease.
Prog. Neurobiol. 83, 131-139 (2007) Review
M. Tabaton and E. Tamagno
The molecular link between betaand gamma-secretase activity on the amyloid beta precursor protein.
Cell Mol. Life Sci. 64, 2211-2218 (2007) Review
B. Van Broeck et al.
Current insights into molecular mechanisms of Alzheimer disease and their implications for therapeutic approaches.
Neurodegener. Dis. 4, 349-365 (2007) Review
L.B. Hersh and D.W. Rogers
Neprilysin and amyloid beta peptide degradation.
Curr. Alzheimer Res. 5, 225-231 (2008) Review
Y. Ohyagi
Intracellular amyloid beta-protein as a therpeutic target for treating Alzheimer’s disease.
Curr. Alzheimer Res. 5, 555-561 (2008) Review
K.A. Bates et al.
Clearance mechanisms of Alzheimer’s amyloid-beta peptide: implications for therapeutic design and diagnostic tests.
Mol. Psychiatry 14, 469-486 (2009) Review
R. Deane et al.
Clearance of amyloid-beta peptide across the blood-brain barrier: implication for therapies in Alzheimer’s disease.
CNS Neurol. Disord. Drug Targets 8, 16-30 (2009) Review
F. Song et al.
Plasma biomarkers for mild cognitive impairment and Alzheimer’s disease.
Brain Res. Rev. 61, 69-80 (2009) Review
E. Bruno et al.
Lack of interaction between LRP1 and A2M polymorphisms for the risk of Alzheimer disease.
Neurosci. Lett. 482, 112-116 (2010)
H.J. Garringer et al.
Modeling familial British and Danish dementia.
Brain Struct. Funct. 214, 235-244 (2010) Review
A. Kitamura and H. Kubota
Amyloid oligomers: dynamics and toxicity in the cytosol and nucleus.
FEBS J. 277, 1369-1379 (2010) Review
B. Liang et al.
Calpain activation promotes BACE1 expression, amyloid precursor protein processing, and amyloid plaque formation in a transgenic mouse model of Alzheimer disease.
J. Biol. Chem. 285, 27737-27744 (2010)
C.J. Lin et al.
Cu(II) interaction with amyloid-beta peptide: a review of neuroactive mechanisms in AD brains.
Brain Res. Bull. 82, 235-242 (2010) Review
M.L. Moro et al.
Alzheimer’s disease and amyloid beta-peptide deposition in the brain: a matter of ‘aging’?
Biochem. Soc. Trans. 38, 539-544 (2010) Review
K. Murakami et al.
The turn formation at positions 22 and 23 in the 42-mer amyloid beta peptide: the emerging role in the pathogenesis of Alzheimer’s disease.
Geriatr. Gerontol. Int. 10 Suppl 1, S169-S179 (2010)
M.P. Murphy and H. LeVine 3rd
Alzheimer’s disease and the amyloidbeta peptide.
J. Alzheimers Dis. 19, 311-323 (2010) Review
J.F. Quinn et al.
A copper-lowering strategy attenuates amyloid pathology in a transgenic mouse model of Alzheimer’s disease.
J. Alzheimers Dis. 21, 903-914 (2010)
D.R. Thal et al.
Capillary cerebral amyloid angiopathy identifies a distinct APOE epsilon4-associated subtype of sporadic Alzheimer’s disease.
Acta Neuropathol. 120, 169-183 (2010)
N. Venketasubramanian et al.
Interethnic differences in dementia epidemiology: global and Asia-Pacific perspectives.
Dement. Geriatr. Cogn. Disord. 30, 492-498 (2010) Review
B. Zapala et al.
Humanins, the neuroprotective and cytoprotective peptides with antiapoptotic and anti-inflammatory properties.
Pharmacol. Rep. 62, 767-777 (2010)
C. Humpel
Identifying and validating biomarkers for Alzheimer’s disease.
Trends Biotechnol. 29, 26-32 (2011)
S. Jawhar et al.
Pyroglutamate amyloid-beta (Abeta): a hatchet man in Alzheimer disease.
J. Biol. Chem. 286, 38825-38832 (2011)
R. Mayeux and N. Schupf
Blood-based biomarkers for Alzheimer’s disease: plasma Abeta40 and Abeta42, and genetic variants.
Neurobiol. Aging 32 Suppl 1, S10-S19 (2011)
B. Vincent and P. Govitrapong
Activation of the alpha-secretase processing of AbetaPP as a therapeutic approach in Alzheimer’s disease.
J. Alzheimers Dis. 24 Suppl 2, 75-94 (2011)
W.T. Chen et al.
Amyloid-beta (Abeta) D7H mutation increases oligomeric Abeta42 and alters properties of Abeta-zinc/copper assemblies.
PLoS ONE 7, e35807 (2012)
W. Danysz and C.G. Parsons
Alzheimer’s disease, beta-amyloid, glutamate, NMDA receptors and memantine – searching for the connections.
Br. J. Pharmacol. 167, 324-352 (2012)
R. Epis et al.
Alpha, beta-and gamma-secretases in Alzheimer’s disease.
Front. Biosci. (Schol. Ed.) 4, 1126-1150 (2012)
B.L. Kagan et al.
Antimicrobial properties of amyloid peptides.
Mol. Pharm. 9, 708-717 (2012)
C.B. Pocernich and D.A. Butterfield
Elevation of glutathione as a therapeutic strategy in Alzheimer disease.
Biochim. Biophys. Acta 1822, 625-630 (2012)
N.E. Pryor et al.
Unraveling the early events of amyloid-beta protein (Abeta) aggregation: Techniques for the determination of Abeta aggregate size.
Int. J. Mol. Sci. 13, 3038-3072 (2012)
N. Sun et al.
A survey of peptides with effective therapeutic potential in Alzheimer’s disease rodent models or in human clinical studies.
Mini Rev. Med. Chem. 12, 388-398 (2012) Review
L.N. Zhao et al.
The toxicity of amyloid beta oligomers.
Int. J. Mol. Sci. 13, 7303-7327 (2012)
J.L. Crimins et al.
The intersection of amyloid beta and tau in glutamatergic synaptic dysfunction and collapse in Alzheimer’s disease.
Ageing Res. Rev. 12, 757-763 (2013)
R. Perez-Garmendia and G. Gevorkian
Pyroglutamate-Modified Amyloid Beta Peptides: Emerging Targets for Alzheimer s Disease Immunotherapy.
Curr. Neuropharmacol. 11, 491-498 (2013)
E. Drolle et al.
Atomic force microscopy to study molecular mechanisms of amyloid fibril formation and toxicity in Alzheimer’s disease.
Drug. Metab. Rev. 46, 207-223 (2014)
R.D. Johnson et al.
Structural evolution and membrane interactions of Alzheimer’s amyloidbeta peptide oligomers: new knowledge from single-molecule fluorescence studies.
Protein Sci. 23, 869-883(2014)
M.P. Kummer and M.T. Heneka
Truncated and modified amyloidbeta species.
Alzheimers Res. Ther. 6, 28 (2014)
S. Schedin-Weiss et al.
The role of protein glycosylation in Alzheimer disease.
FEBS J. 281, 46-62 (2014)
A. Martorana et al.
Cerebrospinal Fluid Aβ42 Levels: When Physiological Become Pathological State.
CNS Neurosci. Ther. 21, 921-925 (2015)
L. Montoliu-Gaya and S. Villegas
Protein structures in Alzheimer’s disease: The basis for rationale therapeutic design.
Arch. Biochem. Biophys. 588, 1-14 (2015)
M.R. Nichols et al.
Biophysical comparison of soluble amyloid-beta(1-42) protofibrils, oligomers, and protofilaments.
Biochemistry 54, 2193-2204 (2015)
D. Puzzo et al.
The keystone of Alzheimer pathogenesis might be sought in Abeta physiology.
Neuroscience 307, 26-36 (2015)
H.H. Jarosz-Griffiths et al.
Amyloid-beta Receptors: The Good, the Bad, and the Prion Protein.
J. Biol. Chem. 291, 3174-3183 (2016)
F.Z. Javaid et al.
Visual and Ocular Manifestations of Alzheimer’s Disease and Their Use as Biomarkers for Diagnosis and Progression.
Front. Neurol. 7, 55 (2016)
T. Mohamed et al.
Amyloid cascade in Alzheimer’s disease: Recent advances in medicinal chemistry.
Eur. J. Med. Chem. 113, 258-272 (2016)
Z.X. Wang et al.
The Essential Role of Soluble Abeta Oligomers in Alzheimer’s Disease.
Mol. Neurobiol. 53, 1905-1924 (2016)
X. Zhou et al.
An overview on therapeutics attenuating amyloid beta level in Alzheimer’s disease: targeting neurotransmission, inflammation, oxidative stress and enhanced cholesterol levels.
Am. J. Transl. Res. 8, 246-269 (2016) Review
Y. Zhou et al.
Detection of Abeta Monomers and Oligomers: Early Diagnosis of Alzheimer’s Disease.
Chem. Asian J. 11, 805-817 (2016)